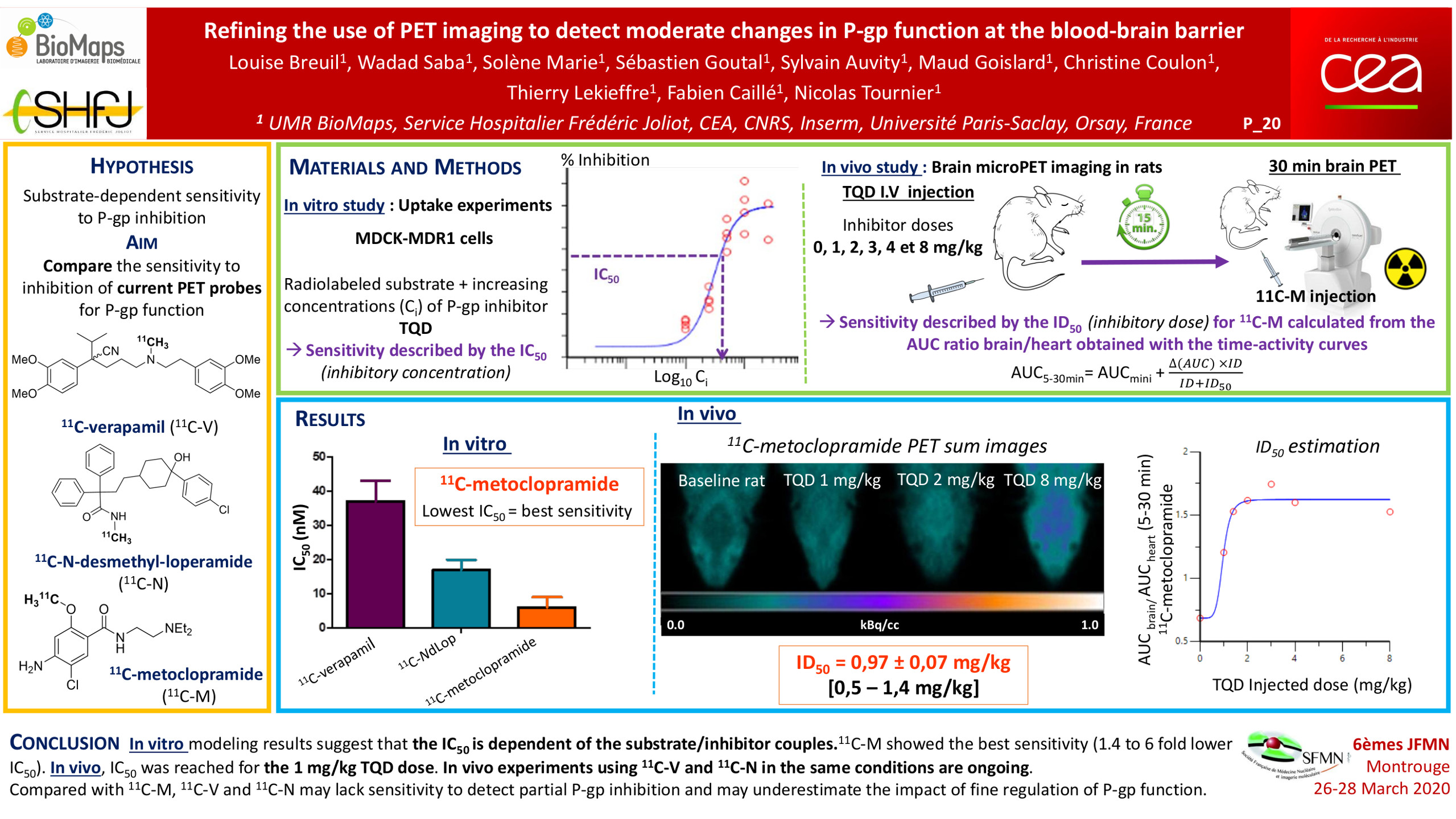
In the clinical practice, complete repression of P-glycoprotein (P-gp) function at blood-brain barrier (BBB) is not likely to occur. However, partial inhibition/deficit is suspected in many pathophysiological situations. PET imaging using radiolabeled substrates of P-glycoprotein (P-gp) is the most advanced technique to study P-gp function at the human BBB. However, the sensitivity of current radiolabeled P-gp substrates to detect moderate changes in P-gp function at the BBB remained to be compared. In vitro and in vivo studies were conducted to compare 11C-verapamil (11C-V), 11C-N-desmethyl-loperamide (11C-N) and 11C-metoclopramide (11C-M). In vitro uptake assays were performed in MDCKII-hMDR1 cells to compare the transport capacity and the 50% inhibitory concentration (IC50) toward the P-gp inhibitors tariquidar (TQD) and cyclosporine A (CsA) for each radiolabeled substrate. In vivo, increasing i.v. doses of TQD on the brain distribution of 11C-M were first studied by microPET imaging in rats. In vitro modeling results suggest that the transport capacity and IC50 are highly dependent of the substrate/inhibitor couples. With both inhibitors, 11C-M showed the best sensitivity (1.4 to 6 fold lower IC50) and the lowest transport capacity. In vivo, IC50 was reached for the 1 mg/kg TQD dose. In vivo experiments using 11C-V and 11C-N in the same conditions are ongoing. Compared with 11C-M, avid P-gp substrates (11C-V and 11C-N) may lack sensitivity to detect partial P-gp inhibition/deficit and may underestimate the impact of fine regulation of P-gp function at the human BBB.


